Die Ausgaben der gesetzlichen Krankenkassen für Arzneimittel steigen seit Jahren trotz aller gesetzgeberischen Versuche, Grenzen zu ziehen und die Kostenentwicklung einzudämmen. Mit dem Arzneimittelmarktneuordnungsgesetz (AMNOG) aus dem Jahr 2010 wollte der Gesetzgeber diesen Trend endlich beenden. Zur Sicherstellung einer zweckmäßigen, qualitativ hochwertigen und wirtschaftlichen Arzneimittelversorgung sowie zur Gewährleistung der finanziellen Stabilität des deutschen Gesundheitssystems in Deutschland wurden pharmazeutische Unternehmer von nun an verpflichtet, für jedes ab dem 1. Januar 2011 in den deutschen Markt eingeführte erstattungsfähige Arzneimittel mit einem neuen Wirkstoff den Zusatznutzen gegenüber einer zweckmäßigen Vergleichstherapie nachzuweisen (§ 35a SGB V). Auf Basis des Beschlusses des G-BA über die Nutzenbewertung verhandeln der GKV-Spitzenverband und der pharmazeutische Unternehmer für das Arzneimittel den sog. „Erstattungsbetrag“ (§ 130b SGB V), es sei denn, der G-BA hat das Arzneimittel einer Festbetragsgruppe zugeordnet. Die Verhandlung des Erstattungsbetrages richtet sich maßgeblich nach dem Zusatznutzen. Für Arzneimittel mit Zusatznutzen gelten andere Verhandlungs- und Preisvorgaben als für Arzneimittel ohne Zusatznutzen. Seit dem GKV Finanzstabilisierungsgesetz (GKV-FinStG) aus dem Jahr 2022 ist ebenfalls der Bestand oder Wegfall des Patent – und Unterlagenschutzes der zweckmäßigen Vergleichstherapie relevant (sog. „Leitplanken“); auch die Aussagewahrscheinlichkeit ist seither ausdrücklich als Preisfindungsaspekt gesetzlich verankert. Mit dem Medizinforschungsgesetz (MFG) wurde ab dem 1. Januar 2025 die Option eingeführt, den verhandelten Erstattungsbetrag nicht ins öffentliche Preis– und Produktverzeichnis melden zu müssen. Es besteht damit die Möglichkeit für den pharmazeutischen Unternehmer einen vertraulichen Erstattungsbetrag, einen Geheimpreis, zu verlangen, sofern er eine Forschungstätigkeit in Deutschland nachweist. Des Weiteren wurden Ausnahmen von den Verhandlungsvorgaben des GKV-FinStG eingeführt (die sog. „Leitplankenausnahme“). Generell gilt: Können sich die Parteien nicht einigen, setzt eine Schiedsstelle die offenen Vertragsinhalte per Schiedsspruch fest. Der Erstattungsbetrag gilt ab dem 7. Monat nach dem erstmaligen Inverkehrbringen des Arzneimittels sowie nach Zulassung neuer Anwendungsgebiete als neuer Abgabepreis für die gesetzlich Versicherten sowie Privatversicherte und Selbstzahler. Für Krankenhäuser gilt er im Einkauf als Höchstpreis.
Grundgedanken eines Systems im Wandel
Seit seiner Einführung wird das AMNOG als „lernendes System“ charakterisiert; dies spiegelt sich darin wider, dass sich der rechtliche Rahmen des „AMNOG-Systems“ im ständigen Wandel befindet.
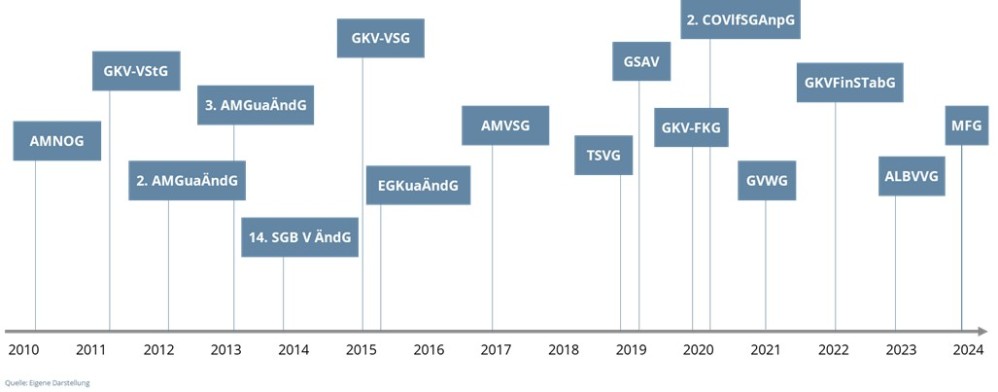
Seit Inkrafttreten des AMNOG am 1. Januar 2011 wurde allein der den Verhandlungen zugrundeliegende § 130b SGB V bis 31.12.2024 in bisher sechzehn Gesetzen, d. h. im Schnitt mehr als einmal jährlich (1,14 x pro Jahr) verändert (Abbildung 1).